
Source RAPS
Addressing manufacturing controls for the cell and gene therapy industry, this article discusses criticality of establishing Chemistry Manufacturing Controls (CMC) Readiness, Critical Quality Attributes (CQAs) and Critical Process Parameters (CPPs) for cell and gene therapy products. The author suggests manufacturers need sound drug development and manufacturing facility plans covering a product’s lifecycle and should establish sustainable manufacturing control strategies for each product class. The author also identifies specific guidance and support materials from a number of governmental and commercial organizations to assist in developing products that can be approved by regulators and potentially commercially successful.
Introduction
The term “regenerative medicine” defines treatment modalities for assisting, stimulating or replacing the body’s own reparative properties using cell, gene therapy or tissue engineering approaches. FDA has approved a number of unique regenerative medicine products, most of which, arguably, have not been commercially successful in either the US or globally. While it is assumed that regulatory approval often leads to commercial success, this has not been the overwhelming trend. The major reasons for such little success are unmistakable and discussed briefly in this article. In general, various types of products evaluated under the wide regenerative medicine umbrella include cell therapy products, gene modified cells for ex-vivo therapy, in-vivo gene therapy products, combination products and others. Each product class has been shown to have unique attributes, manufacturing process and control requirements. Some of these products are “living drugs” that mediate their action by evolving and changing continuously in-vivo. Understanding of how these products work and how these products could be consistently manufactured at commercial scale must be an essential part of the product development cycle. However, FDA currently may not have sufficient resources to exercise appropriate quality oversight of these products, both before and after approval. This lack of resources, combined with an increasing number of Investigational New Drugs (INDs) and postapproval regulatory oversight responsibilities in this area, coupled with FDA resource planning and staffing challenges, could potentially exacerbate the gap between regulatory approval and commercial success.
Background
What is Chemistry Manufacturing Controls (CMC) readiness?
Historically, FDA has set the bar very high regarding the CMC readiness of a licensed product. The licensure requires applicants to demonstrate the final drug product can be manufactured consistently in accordance with a predetermined set of release specifications and the product’s quality attributes over the shelf-life. Regarding cell and gene therapy products, the agency has shown flexibility as to what should constitute reasonable and scientifically justified quality controls and metrics. Yet, manufacturers of some of the approved product in the market struggle to meet their own proposed release specifications after approval.1 Manufacturers also struggle with how to procure critical materials and do so in a sustainable, cost effective manner. This is not a sustainable strategy for commercial success and has a great impact on patients who could benefit from these advanced therapies.
Essential Part of Establishing Manufacturing Controls
What is the product?
It has been noted that, in the case of biologics, the “process is the product,” but this is not the whole story. Looking at the diverse array of products, one can realize that each product has unique attributes. Considering the diverse characteristics of these products [summarized in Tables 1 and 2) is an essential part of establishing manufacturing control, as there is no “turn-key solution,” i.e., one solution for all the problems, available. In reality, manufacturers must identify several “turn-key solutions” or tools that could enable consistent manufacturing of high quality products for one product type at a time.
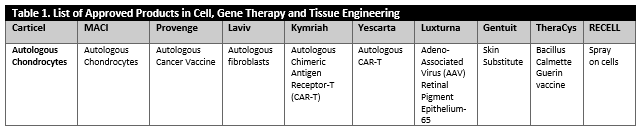
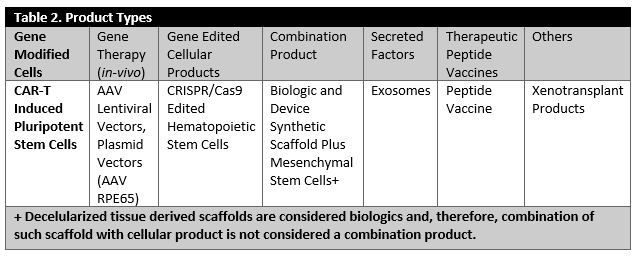
Tables 1 and 2 describe an overview of a diverse array of products which are approved and are currently under investigations. These products include cellular products, gene modified cellular product, viral and non-viral vector products (in-vivo gene therapy), combinations products, ex-vivo gene edited cellular products and peptide based cancer vaccines or products that consist of secreted and extracted biological entities.
Autologous Versus “off the Shelf”
As outlined in Table 3, the challenges in establishing manufacturing controls are quite different for autologous products versus “off-the-shelf” products, which are used either without any Human Leukocyte Antigen (HLA) matching or very minimal matching. There are some common manufacturing challenges shared by both autologous and allogeneic products. These include mode of action, material qualification, establishing meaningful specifications, manufacturing facility design and controls, material shipping and handling and managing manufacturing changes. However, autologous products have unique characteristics needing to be considered more rigorously. These characteristics include manufacturing logistics, scheduling and inherent variability in the product based on the nature of the starting material and scale-out considerations. The specific concerns for off-the-shelf products, on the other hand, include donor eligibility, qualification, reproducibility, stability of the master cell bank and scale-up considerations.
NOTE: in autologous therapies, the patient is both donor of starting biological materials used to manufacture the final product and the recipient. Allogeneic refers to product derived from one donor and given to unrelated recipients.
Figure 1 provides a summary of points to consider at a very high level based on the nature of the product. While there are many common challenges (for example, major considerations of manufacturability, safety, cost and consistency), there are also product-specific challenges for each product type identified (Table 3).
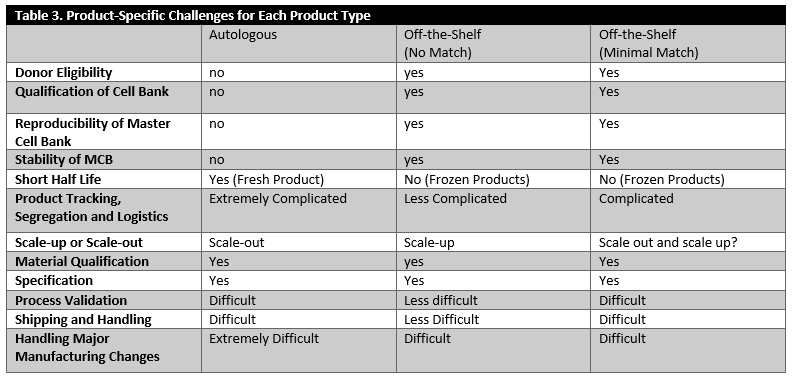
Figure 1. Product Specific Major Manufacturing Challenges

Leveraging Product Knowledge
The process of acquiring in-depth product knowledge for well-characterized biologics and small molecules is well established and includes a development approach yielding in-depth understanding of the Critical Quality Attributes (CQAs) of the product and the need to better define critical manufacturing steps and Critical Process Parameters (CPPs). However, this approach has, for a variety of reasons, yet to be fully adopted by the cell and gene therapy industry. Some of these reasons for this are related to the challenging technical issues and inherent biological limitations for the products developed and manufactured in this space. Understanding product CQAs/CPPs is perhaps the most critical aspect of establishing a suitable manufacturing process as well as establishing controls for assuring product quality and consistency.
For these products, CQAs are the biological and/or molecular characteristics of a critical material, drug substance or drug product which are informative in measuring product quality, safety and efficacy. Moreover, identifying and understanding the product specific and relevant CQAs are extremely helpful in the development of meaningful specifications, which is yet another part of establishing and ensuring manufacturing quality and consistency. FDA recognizes that identifying product specific and clinically relevant CQAs is extremely complicated. Accordingly, the agency encourages a systematic approach involving several steps. These steps include:
- Starting with the identification of several candidate CQAs based on the Mode of Action (MOA) of each product and the development of qualified assays to measure such candidate CQAs. The knowledge gathered during the product development cycle form a scientific basis for establishing meaningful specifications.
- In addition, there should be a systematic approach to correlate CQAs with product quality and clinical outcomes forms the basis for establishing the biological and clinical relevance of each candidate CQA.
- Through a highly systematic and iterative process, it is possible to identify potential CQAs which are found to be potentially clinically relevant (Figure 2).
Figure 2. Essential Steps for Identifying Product Specific CQAs and Their Clinical Relevance

Critical Process Parameters (CPP)
CPPs are independent process parameters most likely affected by critical quality attributes. It is important to note that CPPs cannot be identified in the absence of CQAs. However, those process parameters not necessarily linked to CQAs could be identified as Key Process Parameter (KPP). In the absence of CPPs, or partial understanding of CPPs, full knowledge of KPPs could become equally important.
Understanding CQAs is an Essential Tool for Establishing Product Potency
Regenerative medicine product manufacturers are required to establish pre-determined, justified release specifications for potency, which is arguably the most critical aspect of establishing manufacturing control and consistency. Although FDA regulations regarding potency appear to be flexible, early stage development of several candidate potency assays, linked to the product CQAs, should be considered an essential part of establishing manufacturing control. Although they are not necessarily required, potency assays ideally should be validated for adherence to Good Laboratory Practice (GLPs) and current Good Manufacturing Practice (CGMPs) when used during trials to determine product efficacy.
Understanding CQAs and CPPs is an Essential Tool for Establishing Process Optimization
As additional knowledge is gained, in the interest of making a more consistent, quality product, changes to the manufacturing process/product become an inevitable part of process development. Risking manufacturing changes late in the clinical trials could potentially impact the product’s critical characteristics. For this reason, manufacturers are encouraged to introduce major manufacturing changes as early as possible during the product development lifecycle and demonstrate the products, both pre-and post-change, are highly similar.
In reality, and due to a variety of business and logistical reasons, manufacturers often introduce major manufacturing changes late in the product development lifecycle. Some of these common changes include “scale-up” and introduction of process automation.
Foreign regulatory authorities, as well as FDA have broadly adopted the requirements defined in ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process for establishing and demonstrating product comparability, postchange. A comparability study is defined as a prospective study protocol designed to demonstrate that two products are comparable before and after a given change.2 In some cases, FDA may ask an IND sponsor or Biologics License Applicant (BLA) to submit their comparability study for assessment and review prior to data collection and analysis.
In ICH Q5E, comparability is defined as a
Fortunately, the minimal elements of a good comparability study for this emerging product class have been defined in several public presentations by FDA’s Center for Biologics Evaluation and Research (CBER). Based on available information, the following are considered by FDA to be critical elements of a good, prospective comparability study for cell and gene therapy products.4
- description of the proposed change(s)
- risk assessment
- rationale for the proposed change(s)
- comparability study design for the proposed change(s)
- comparative assessment of quality attributes before and after change (side-by-side comparison is preferred)
- justification for well-defined pre-specified acceptance criteria for establishing comparability
- detailed analytical procedures, sampling plan, statistical methods and analysis
- reporting commitment
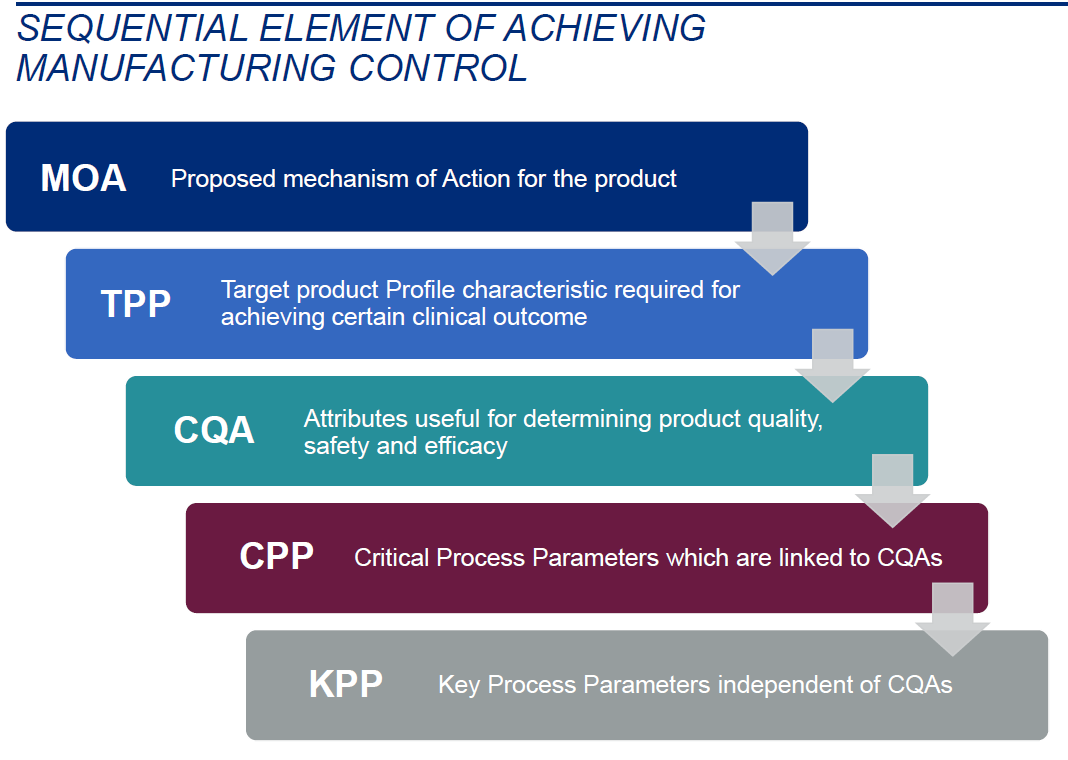
Establishment of Appropriate Manufacturing Controls
Establishing appropriate manufacturing controls means BLA applicants have established sufficient CMC readiness to manufacture the final drug product consistently and in accordance with well-defined, pre-determined specifications. If appropriate manufacturing controls are not fully established prior to approval, manufacturers will be at-risk for market failure and of not meeting patient needs due to an increasing complexity of manufacturing at commercial scale, postapproval. Sequential elements of establishing appropriate manufacturing controls (Figure 3) rely on a manufacturer’s full understanding of their product as well as the development of manufacturing processes that employ a robust understanding of the potential failure modes.5
Figure 3. Sequential Element of Achieving Manufacturing Control
Manufacturing Platforms Require Further Development
Manufacturing in the cell and gene therapy space is undergoing significant change as new technologies and devices are developed to perform specific manufacturing steps in functionally closed systems. These manufacturing or automated systems are expected to streamline manufacturing steps and also may allow for more flexible manufacturing environments (Figure 4). However, these technologies are at the early stages of incorporation and require further improvement in automation and software design, development and reliability. As they are subject to routine failure, these platforms also remain somewhat dependent on interventions by trained operators. An equally important aspect of manufacturing improvements is the development of a more standardized method for collecting the starting material. While this area includes only a handful of industrial players, standardization of the collection processes remains a challenge to be overcome.
Figure 4. Examples of the Existing Platforms Used in Manufacturing Cell and Gene Therapy Products

To address and overcome manufacturing challenges currently facing the cell and gene therapy industry, FDA, the National Institute of Standards (NIST), the US Department of Defense (DOD) and the National Institutes of Health (NIH) have encouraged the development of new public/private consortiums, such as the NIST-supported National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL), the Advanced Regenerative Medicine Institute (ARMI), supported by the DOD and other initiatives supported by NIH.6
While the focus of ARMI is on the development of tissue engineering products and 3D printing supported by the DOD, NIIMBLrecently published two detailed documents covering the “roadmap” for gene therapy, vaccines and bispecific products.
In parallel, FDA has initiated several programs to enhance manufacturing technologies, including the recent announcement of an R01 grant for advanced manufacturing (FDA/CBER Enhancing Innovations in Emerging Technologies for Advanced Manufacturing of Complex Biologic Products (R01).
Finally, continuous manufacturing is considered to be a potentially helpful solution in the development of biotech-like products, such as viral vector production.
When considering an innovative manufacturing or analytical approach for these types of products, early manufacturer engagement with FDA is encouraged prior to formal submission.
Standard Development in Regenerative Medicine is Lagging
The development of tools for standardization of product manufacturing is another area of focus. To accomplish the development of physical and performance standards, several initiatives have been initiated by consensus from standard development bodies, such as the International Society for Computed Tomography (ISCT), the International Organization for Standardization (ISO), the American Society for Testing and Materials (ASTM) and non-consensus organization, such as US Pharmacopeia (USP).
FDA also has been proactive in publishing draft guidance documents, which should be helpful guiding the thought process in the industry.7
A summary of key players involved in standard development, which include the development of physical and performance standards by consensus and non-consensus standard development bodies, includes:
- Standard Coordinating Body (SCB) is a not for profit organization began as an initiative of the Alliance for Regenerative Medicine (ARM). In September 2016, the National Institute of Standards and Technology (NIST) and SCB established a Memorandum of Understanding (MOU).
- Standard Development Bodies (Consensus and Non-Consensus)
- ISCT, ASTM, ISO, USP
- National Institute of Standards and Technologies (NIST)
Manufacturing Facilities Considerations
The current state and design of facilities where drug products for clinical trials are manufactured often determines the quality of the final drug product. In the US, unlike Europe, facilities are not necessarily inspected for compliance with CGMPs during Phase I trials, but are verified at the time of the pre-license inspection. For business reasons, companies often start by manufacturing their products in “less than ideal” environments. For example, sometimes they may use a regular research laboratory with a Biological Safety Lab 2 biological cabinet or a room not intended to be a manufacturing suite for clinical grade products (Stage I). As the product shows some promise, sponsors may move their manufacturing to a more suitable environment which is more controlled and classified (Stage II). Finally, if the product is a success in Phase III, the third stage of facility upgrade may involve building a new manufacturing facility or contracting with a CGMP-licensed manufacturer.
It is important to note that facility and cleanroom environmental controls and monitoring are critical aspects of manufacturing a quality product under CGMPs. This is particularly true since many cell and gene therapy products are not terminally sterilized and/or sterile filtered, with the result that quality tests could potentially be performed with samples not necessarily representative of the final drug product. Additionally, the facility’s environment remains as the major source of contamination in the final drug product. In such situations, it is particularly important to manufacture clinical grade products in an environment deemed to be suitable for the product type and based on the analysis of critical manufacturing and processing steps which could potentially place products in direct contact with the environment.
Manufacturers often “scramble” during late stages of clinical trials to get ready for commercial manufacturing by dedicating considerable resources to building new, state-of-the-art manufacturing facilities. Planning for success early on allows for streamlining the process for finding an appropriate manufacturing environment flexible enough to accommodate further expansion depending on the intended scale of commercial manufacturing achieved through either “scale-up” or “scale-out.” While a scale-up strategy may not require a large expansion, a scale-out of manufacturing, involving replication of manufacturing unit operations, definitely will. In view of these challenges, which are an inevitable part of the product development cycle, developing a CGMP facility strategy early on is strongly recommended to address manufacturing facility requirements.
As manufacturers plan for success they may want to consider the following points when designing or selecting a facility for biologic product manufacturing:
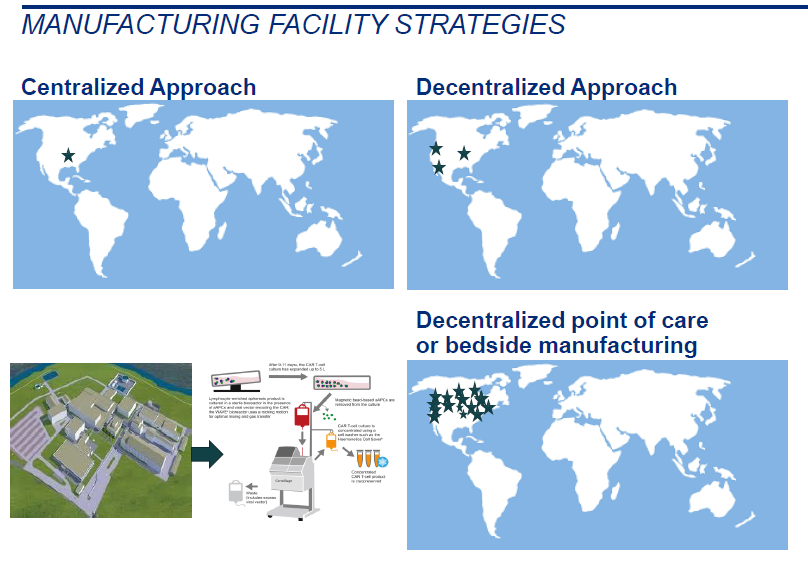
- What is a suitable model for manufacturing the final drug product? Centralized or decentralized models versus contract manufacturing by a third party. If decentralized model is followed, is it feasible to perform manufacturing at the near patient manufacturing sites? Is it possible to manufacture the drug product at point of care? (Figure 5)
- What is the appropriate and flexible environment for manufacturing of the product based on the current state of manufacturing?
- Can they move to manufacturing platforms earlier which are less open to the environment and more amenable to aseptic sampling?
- If not, what are the available manufacturing platforms that are suitable for automation of the process? Are there any existing, functionally-closed platforms that allow manufacturers to manufacture products requiring less environmental control and monitoring?
- Can isolators be used as part of the manufacturing process?
- What is the plan for commercial manufacturing scale-out or scale-up considerations?
- Will a contract manufacturer be used and if so, is an appropriate quality agreement in place to ensure robust manufacture of the commercial product?
- Has the plan about commercial scale manufacturing been discussed with the regulatory agency?
- What are implications of changing manufacturing sites from one site to another in US or moving the manufacturing site from outside of US to US during critical phases of clinical studies?
Figure 5. Differences Between the Centralized, Decentralized and Near Patient Manufacturing Schematically
Conclusion
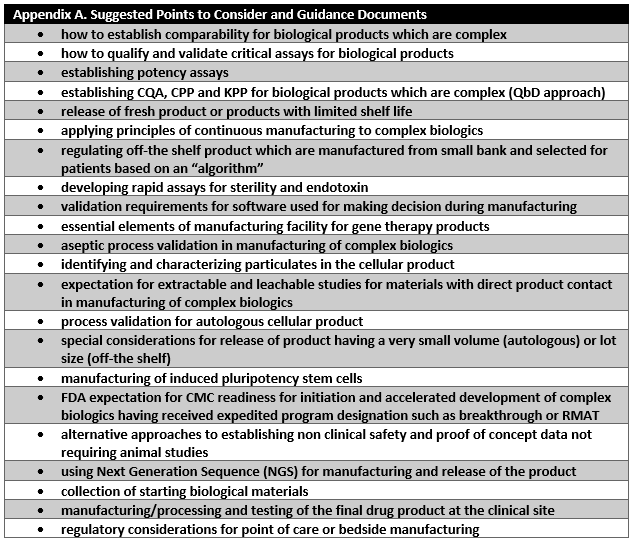
CMC readiness remains one of the major challenges in the expedited development of cell and gene therapy products. Manufacturers and regulators need to make sure that a reasonable degree of manufacturing control is demonstrated by BLA applicants prior to approval. Manufacturers need to lay out a sound development and facility plan covering the product’s lifecycle, including postapproval considerations. FDA, in collaboration with industry, should develop consensus and quality standards for publishing in the form of guidance documents and reasonable and appropriate points to consider for establishing sustainable manufacturing control strategies for each product class. Appendix A lists suggested points to consider that FDA could develop to address key bottlenecks identified in this article for development and approval of cell and gene therapy products.
Recommended Reading
- “Benefits of Identifying Critical Quality Attributes, and Correlating the CQAs with Clinical Outcomes for Biologic Products.” https://regulatory.parexel.com/regulatory-blog/benefits-of-identifying-critical-quality-attributes-and-correlating-the-cqas-with-clinical-outcomes-for-biologic-products. Accessed 17 April 2019.
- “Points to Consider for Establishing Biotechnological/Biological Product Comparability. https://regulatory.parexel.com/regulatory-blog/points-to-consider-for-establishing-biotechnological-biological-product-comparability. Accessed 17 April 2019.
- “Points to Consider When Referencing a Master File in FDA Regulatory Submissions (IND, BLA, NDA).” https://regulatory.parexel.com/regulatory-blog/points-to-consider-when-referencing-a-master-file-in-fda-regulatory-submissions-ind-bla-nda. Accessed 17 April 2019.
- “Points to Consider When Designing a Biologics Manufacturing Facility, Planning for Success Early On!” https://regulatory.parexel.com/regulatory-blog/points-to-consider-when-designing-a-biologics-manufacturing-facility-planning-for-success-early-on. Accessed 17 April 2019.
- “FDA Compliance Deadline for Stem Cell Clinics Offering Unapproved Products to Public.” https://regulatory.parexel.com/regulatory-blog/fda-compliance-deadline-for-stem-cell-clinics-offering-unapproved-products-to-public. Accessed 17 April 2019.
- “Updates on Regenerative Medicine Advanced Therapy (RMAT) Designations.” https://regulatory.parexel.com/regulatory-blog/updates-on-regenerative-medicine-advanced-therapy-rmat-designations. Accessed 17 April 2019.
- ICH Q2(R1)—Includes Validation of Analytical Procedures. https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm265700.htm. Accessed 17 April 2019.
- ICH Q6—Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Product. https://www.ema.europa.eu/documents/scientific-guideline/ich-q-6-test-procedures-acceptance-criteria-new-drug-substances-new-drug-products-chemical_en.pdf. Accessed 17 April 2019.
- ICH Q8—Pharmaceutical Development.https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf. Accessed 17 April 2019.
- ICH Q9—Quality Risk Management. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf. Accessed 17 April 2019.
- ICH Q10—Pharmaceutical Quality Systems. https://www.fda.gov/downloads/drugs/guidances/ucm073517.pdf. Accessed 17 April 2019.
- Process Validation: General Principles and Practices. https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf. Accessed 17 April 2019.
- Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products.https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/general/ucm590118.pdf. Accessed 17 April 2019.
- Guidance for Industry Comparability Protocols-Chemistry, Manufacturing, and Control Information. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070545.pdf. Accessed 17 April 2019.
- FDA Guidance Concerning Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology-Derived Products.https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122879.htm. Accessed 17 April 2019.
- Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs).https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/xenotransplantation/ucm092705.pdf. Accessed 17 April 2019.
- Guidance “Analytical Procedures and Methods Validation for Drugs and Biologics.https://www.fda.gov/downloads/Drugs/Guidances/UCM386366.pdf. Accessed 17 April 2019.
- CGMPs for Phase I Investigational Drugs. https://www.fda.gov/downloads/drugs/guidances/ucm070273.pdf. Accessed 17 April 2019.
- Potency Tests for Cellular and Gene Therapy Products. https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm243392.pdf. Accessed 17 April 2019.
- Consideration for Early Phase Clinical Trials of Cellular and Gene Therapy Products.https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM564952.pdf. Accessed 17 April 2019.
- Expedited Programs for Regenerative Medicine Therapies for Serious Conditions. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM585414.pdf. Accessed 17 April 2019.
- Formal Meeting between the FDA and Sponsors or Applicants of PDUFA Products. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM590547.pdf. Accessed 17 April 2019.
- INTERACT Meeting Initial Targeted Engagement for Regulatory Advice on CBER Products. https://www.fda.gov/BiologicsBloodVaccines/ResourcesforYou/Industry/ucm611501.htm. Accessed 17 April 2019.
- CMC Information for Human Gene Therapy INDs. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610795.pdf. Accessed 17 April 2019.
- Long Term Follow up After Administration of Human Gene Therapy Products.https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610797.pdf. Accessed 17 April 2019.
- Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retroviruses During Products Manufacturing and Patient Follow up.https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610800.pdf. Accessed 17 April 2019.
- Human Gene Therapy for Rare Diseases.https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610802.pdf. Accessed 17 April 2019.
- PDA Technical Report 81. https://www.cellandgene.com/doc/inside-pda-s-technical-report-no-0001. Accessed 17 April 2019.
References
- Eric Palmer E. “Novartis, Still Struggling With Kymriah Manufacturing, is Providing Some out-of-Spec Doses to Patients who ask.” 18 December 2018. Fierce Pharma. https://www.fiercepharma.com/manufacturing/novartis-still-struggling-kymriah-manufacturing-providing-some-out-spec-doses-to. Accessed 17 April 2019.
- ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process.Current Step 4 Version. 18 November 2004. ICH website. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf. Accessed 17 April 2019.
- Ibid.
- Heidaran M. Cell Therapy Product Manufacturing Considerations. CMC Strategy Forum. 17 July 2017. https://cdn.ymaws.com/www.casss.org/resource/resmgr/cmc_no_am_jul_spkr_slds/2017_CMCS_HeidaranMo. Accessed 17 April 2019.
- Finn T. “Early Stage Manufacturing Considerations for Cell Therapy Products.” CASSS. Cell and Gene Therapy Products. 10 July 2018. https://cdn.ymaws.com/www.casss.org/resource/resmgr/cell&gene_therapy/cgtp_slides/2018_CGTP_FinnThomas.pdf. Accessed 17 April 2019.
- Regenerative Medicine Innovation Project. National Institutes of Health (NIH) website. https://www.nih.gov/research-training/medical-research-initiatives/rmi. Accessed 17 April 2019.
- Standards Development and the Use of Standards in Regulatory Submissions Reviewed in the Center for Biologics Evaluation and Research Guidance for Industry. March 2019. FDA website. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/General/UCM589416.pdf. Accessed 17 April 2019.

Leave a Reply